
Processus d’approbation réglementaire des dispositifs médicaux aux États-Unis : La différence entre 510(k) et PMA
Table des matières
- Qu’est-ce que le 510(k) ?
- Qu’est-ce que l’approbation préalable à la mise sur le marché (PMA) ?
- Pourquoi devons-nous passer par des processus d’approbation tels que 510(k) ou PMA ?
- Documents clés de la FDA
- Quand soumettre un nouveau 510(k) pour une modification d’un dispositif existant ?
- Résumé
Vous envisagez de commercialiser un dispositif médical sur le marché américain ? Il est alors important de comprendre les règles établies par la Food and Drug Administration (FDA).
Avant de pouvoir vendre votre dispositif aux États-Unis, il doit être approuvé par la FDA. Il existe deux voies principales : le processus 510(k) et l’approbation préalable à la mise sur le marché (PMA). Chaque voie d’accès comporte ses propres étapes et exigences, et la bonne dépend principalement du type de dispositif que vous fabriquez et du risque qu’il peut présenter pour les patients ou les utilisateurs.
Dans cet article, nous vous donnons un aperçu clair des deux options, nous vous expliquons en quoi elles diffèrent et nous vous aidons à comprendre quand chacune d’entre elles est utilisée et en quoi consiste la procédure.
Qu’est-ce que le 510(k) ?
Le 510(k), officiellement appelé Premarket Notification, est une procédure d’approbation destinée aux dispositifs médicaux présentant un risque faible à modéré (classes I et II). Dans le cadre de cette procédure, le fabricant doit démontrer que le nouveau dispositif est substantiellement équivalent à un produit déjà approuvé (appelé dispositif prédicat).
Quand est-il utilisé ?
- Lors de l’introduction d’un nouveau produit sur le marché
- Lors d’une modification importante d’un dispositif existant (par exemple, de sa technologie, de son matériel ou de son logiciel)
Qu’est-ce que l’approbation préalable à la mise sur le marché (PMA) ?
La PMA (Pre-Market Approval) est une procédure conçue pour les dispositifs à haut risque (classe III), tels que les stimulateurs cardiaques, les cœurs artificiels ou les implants. Cette procédure est plus exigeante et requiert des essais cliniques, des tests en laboratoire et des preuves prouvant la sécurité et de l’efficacité du dispositif.
Quand est-il utilisé ?
- Lorsqu’il n’existe pas de dispositif similaire (prédicat) sur le marché
- Lorsque le dispositif fait appel à une nouvelle technologie ou présente un risque plus élevé
Pourquoi devons-nous passer par des processus d’approbation tels que 510(k)
ou PMA ?
Tout dispositif médical destiné à être mis sur le marché américain doit être approuvé par la FDA (Food and Drug Administration). Sans cette approbation, le produit ne peut être légalement vendu ou commercialisé aux États-Unis.
La procédure d’approbation vise à garantir :
- Sécurité et efficacité – vérifier que le dispositif ne nuit pas aux patients et qu’il fonctionne comme prévu
- Assurance qualité
- Protection des patients - s’assurer que le dispositif n’introduit pas de risques nouveaux et inattendus.
Qu’est-ce qu’un changement important et quel rôle joue-t-il dans le
processus d’approbation ?
Une fois qu’un dispositif a été approuvé (par exemple, par la procédure 510(k)) et que le fabricant souhaite apporter une modification, il doit évaluer si la modification est “importante”.
- Si la modification n’est pas importante, le dispositif peut continuer à être commercialisé dans le cadre de l’autorisation initiale.
- Si la modification est importante, une nouvelle demande est nécessaire - telle qu’une nouvelle notification 510(k) - car la modification pourrait affecter la sécurité, l’efficacité ou l’utilisation prévue du dispositif.
Exemple de ce qui est ou n’est pas un changement significatif :
Si vous avez un cathéter vasculaire et que vous ne changez que sa couleur, il ne s’agit généralement pas d’une modification importante. En revanche, si vous modifiez le matériau qui entre en contact avec le sang, il s’agit d’une modification importante, car elle peut affecter la biocompatibilité et la sécurité du dispositif.
Documents clés de la FDA
Dans le cadre du processus d’approbation de la FDA, l’agence établit des règles claires sur la manière de procéder en fonction de la classification des risques du dispositif médical. Pour aider les fabricants à s’y retrouver, la FDA fournit des documents d’orientation officiels qui expliquent en détail comment choisir la bonne voie d’approbation et quels sont les documents justificatifs requis.
Vous trouverez ci-dessous des liens vers les deux documents les plus importants que tout fabricant de dispositifs médicaux doit connaître :
1. Décider quand soumettre un 510(k) pour une modification d’un dispositif
existant
https://www.fda.gov/media/99812/download
Que contient ce document ? Ce guide est destiné aux fabricants qui disposent déjà d’un dispositif homologué par la FDA (via la procédure 510(k)) et qui prévoient d’y apporter des modifications. La FDA explique en détail comment évaluer si la modification est suffisamment importante pour nécessiter une nouvelle soumission 510(k).
2. Approbation préalable de mise sur le marché (PMA)
https://www.fda.gov/medical-devices/premarket-submissions/premarket-approval-pma
Que contient ce document ? Cette page web décrit le processus complet de PMA (Pre-Market Approval) - la voie d’approbation la plus stricte de la FDA, requise pour les dispositifs à haut risque (classe III).
Quand soumettre un nouveau 510(k) pour une modification d’un dispositif
existant ?
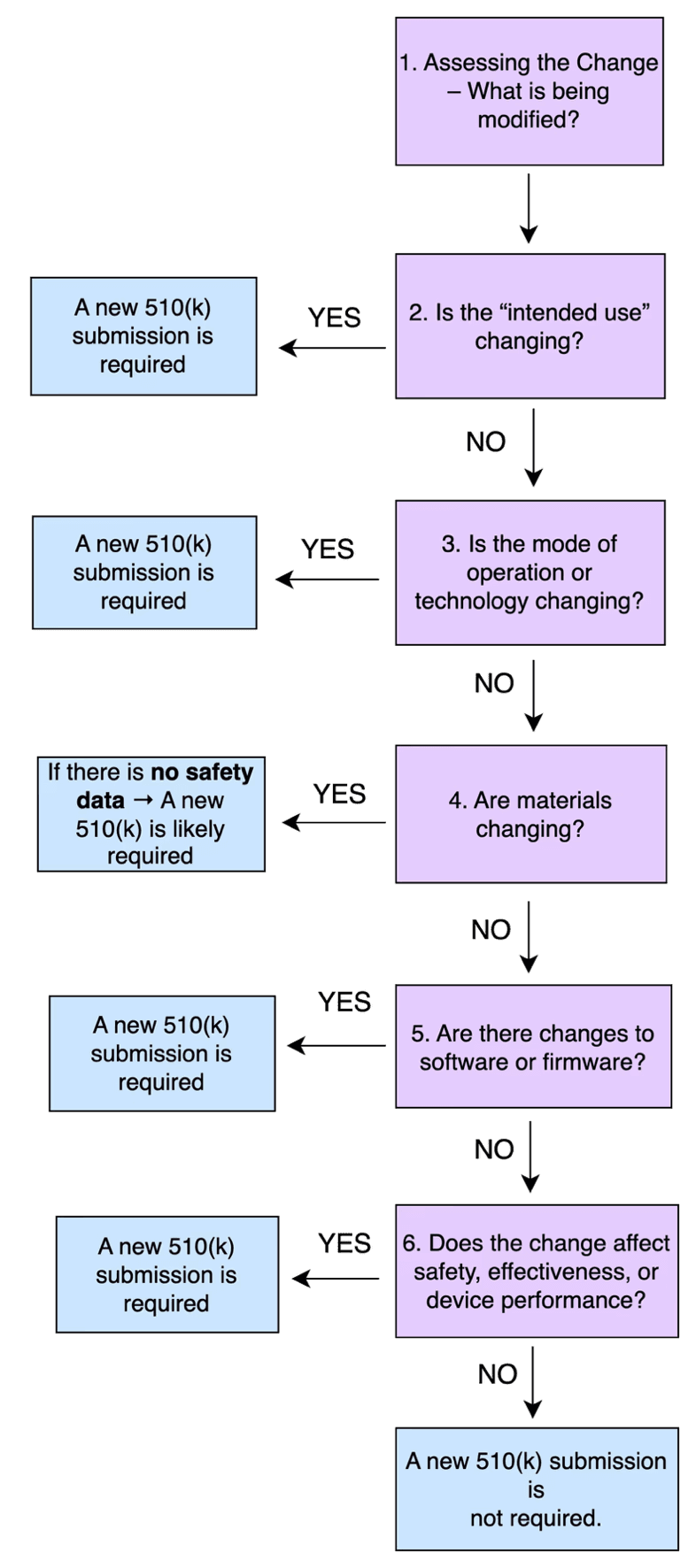
- Si le changement est important → vous devez préparer et soumettre un nouveau 510(k) à la FDA
- Si le changement est insignifiant → , il suffit de documenter le changement en interne et de conserver les enregistrements dans votre système de gestion de la qualité (SGQ). système de gestion de la qualité (SGQ)
- Si vous n’êtes pas sûr → , il est recommandé de consulter un expert en réglementation ou de de demander une réunion de pré- soumission avec la FDA.
Résumé
Si vous envisagez de lancer un dispositif médical sur le marché américain, il est essentiel de déterminer correctement si la procédure 510(k) est suffisante ou si la procédure plus exigeante de l’approbation préalable à la mise sur le marché (PMA) est nécessaire. Chacune de ces voies d’approbation comporte des règles et des critères clairement définis.
En choisissant la mauvaise voie, vous risquez de voir la FDA rejeter votre demande, ce qui peut avoir un impact considérable sur le calendrier de votre projet et le lancement du produit sur le marché.
Comme indiqué précédemment, l’autorisation 510(k) prend généralement de 90 à 180 jours. La procédure d’autorisation de mise sur le marché (PMA) est beaucoup plus longue et peut durer de plusieurs mois à plus d’un an, en fonction de la complexité du dispositif et de la nécessité de disposer de données cliniques.
Si la FDA rejette la demande en raison d’une voie d’accès mal choisie ou d’une documentation insuffisante, il peut être nécessaire de recommencer la procédure.
Ce type d’erreur peut conduire à :
- des retards de plusieurs mois à un an
- l’augmentation des coûts de préparation de la nouvelle documentation
- ou même la perte de débouchés pour le dispositif
C’est pourquoi nous recommandons vivement de travailler avec des experts en réglementation des dispositifs médicaux, qui peuvent vous aider à évaluer la classe de risque du produit, à choisir la stratégie appropriée et à garantir des soumissions précises et complètes à la FDA dès la première tentative.